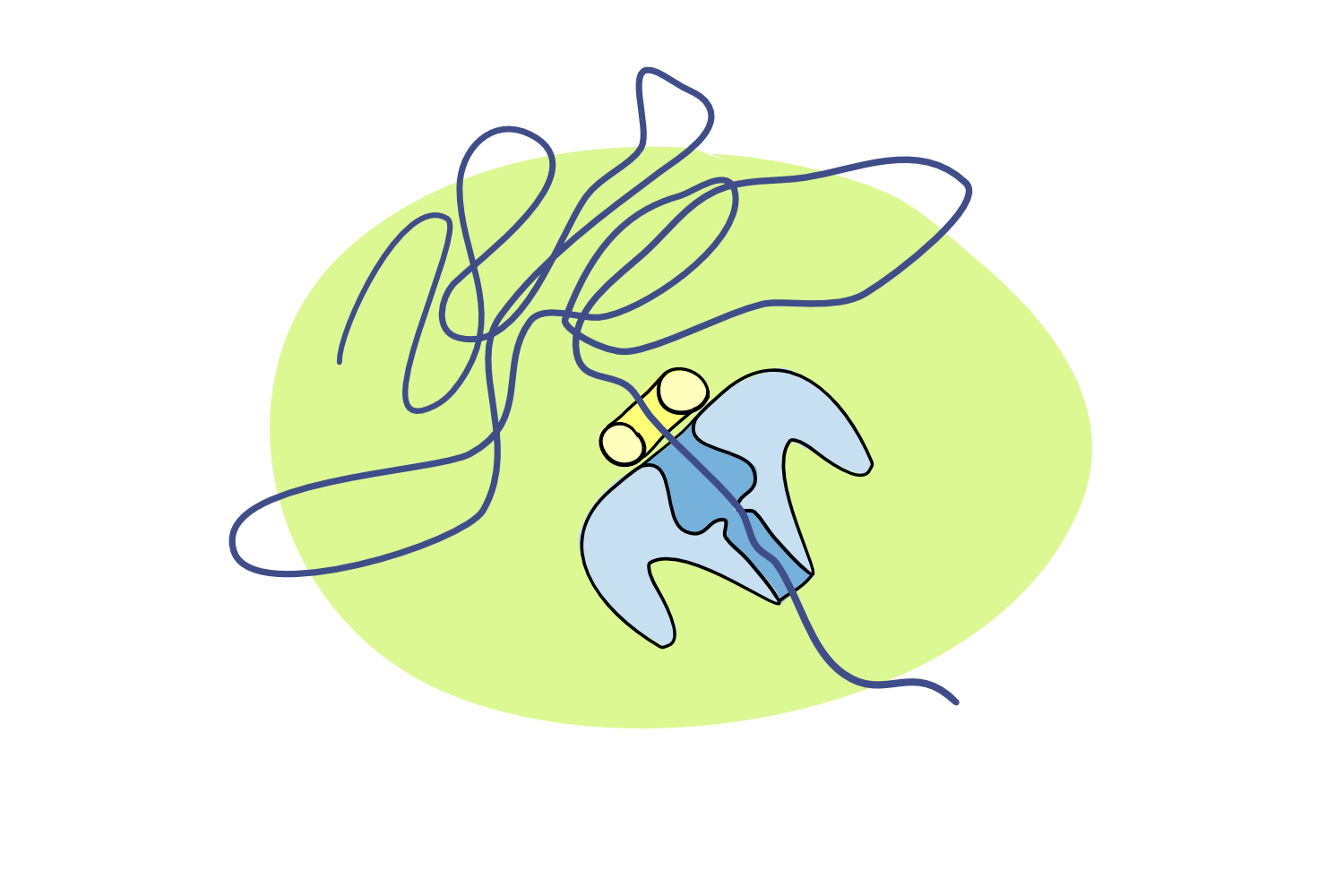
A recent publication describes the extraction of ultra-long DNA. We discuss why the length of a DNA strand is important for science.
I’ve extracted a lot of plant DNA in my time as a scientist. I’ve extracted DNA from thousands, if not tens of thousands of samples, extracted it from different types of plants (mostly arabidopsis and tobacco) and from different plant tissues. I’ve also used a healthy assortment of different protocols to extract my plant DNA- ranging from literal 10 minute-ers, which involve poking at some leaf tissue with a toothpick in buffer, heating it up for a bit and then adding buffer number two, to 20 minute ‘public demonstration’ protocols to protocols that produce nice ‘clean’ DNA and take about 3 hours.
A recent protocol published by Dessireé Zerpa-Catanho and colleagues at the University of Illinois at Urbana-Champaign, describes a painstaking way to extract plant DNA that requires the better part of two days.
The protocol, as detailed here, first involves 2 hours of extraction of plant nuclei – the compartment holding their (nuclear) DNA, then takes a further 22 hours to gently rupture those nuclei to release and precipitate the DNA, and then demands at least another 5 hours to clean up the extracted DNA. To get a single sample of DNA, you need a bunch of time. And did I mention that each sample costs nearly 50 dollars to extract?
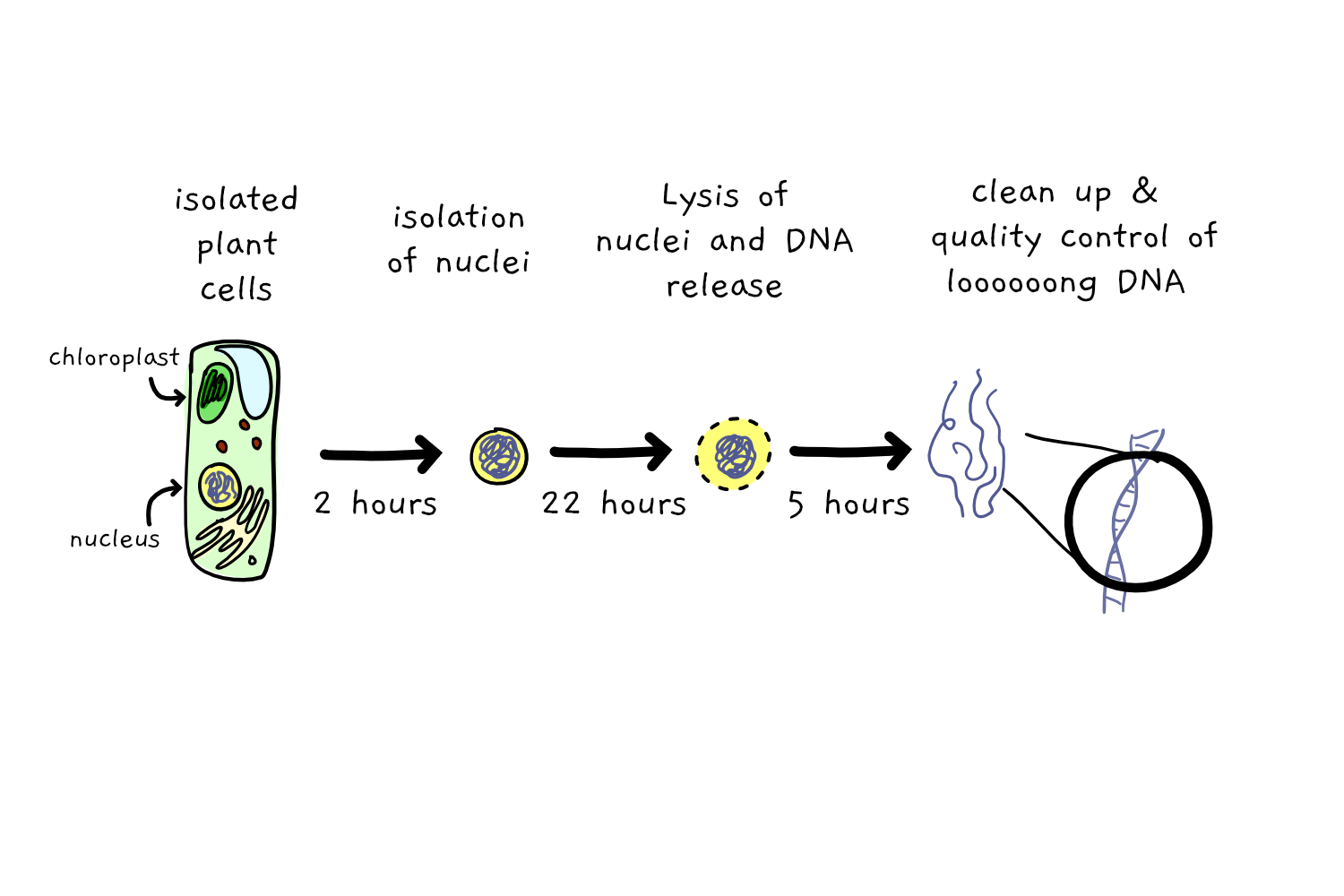
Which seems like a pretty high cost, just to get some DNA.
So the question is – if I can show a 4 year old how to extract plant DNA in about 20 minutes using a ziplock bag, some overripe strawberries, some kitchen soap and salt and just a little bit of alcohol – how is it that a DNA extraction protocol can take so long?
Well as it turns out, not all DNA extractions are created equal. The first consideration is the ‘cleanliness’ of the DNA, indicating how much of what you isolate in your DNA extraction is actual DNA and how much is ‘other stuff’ (RNA and Proteins and sugars OH MY). Depending on what you want to do with your DNA once you have it, the presence of other bits and bobs in the mix can cause pretty big issues. It probably won’t surprise you to know that the good old ‘strawberry bubblebath’ extraction method isn’t a winner on cleanliness. Similarly, those 10 minutes protocols used in the lab can be good for a quick ‘just checking’ PCR, but are fairly useless for anything else.
So the general trend is that for cleaner DNA, we need to use the longer protocols. But even those longer, cleaner protocols can still tend to fall short. Literally.
You see, most DNA extraction methods end up breaking the DNA into short fragments as they pull it out. Nuclear DNA likes to hang out in chromosomes: in the case of Arabidopsis, the approximately 135 mega base pairs (135,000,000) of nuclear genome hang out in just 5 chromosomes ranging from about 20 to about 35 mega bases (20-35,000,000). But when we extract that DNA, the methods to pull out, clean up and even just move around with pipettes, results in short molecules that are generally below 50 kilobases long (50,000).
Generally, short DNA isn’t much of a problem – from a research and technology point of view, we can still do most things we need if the DNA is in smaller pieces. But it is a bit of a problem when it comes to understanding how all of the bits of DNA fit together to make a longer bit of DNA, or, ultimately, a genome. If you have a tonne of short little bits, you’re going to spend a lot of time (virtually) putting them back together in order to get a glimpse of the whole. Something like a puzzle with hundreds of thousands of pieces.
Up until recently, genome sequencing has largely involved two steps. First you sequence all of the DNA – find out what bases are contained in which order. And then you spend a lot of time, and a lot of computer power, trying to work out how all the tiny bits fit together to make a whole.
I should mention that the fact that the DNA bits were short wasn’t originally the fault of DNA extraction methods, but was actually a limit of the sequencing technology itself – which could only read a short stretch of DNA at a time, usually less than 500 bases. So having long pieces of DNA wouldn’t have helped – they would have needed to be cut up before sequencing anyway.
But in the early 2010s, things changed drastically. A new type of sequencing was developed, which allowed DNA to be read in long continuous stretches. The method, called nanopore sequencing, basically involves pulling a thread of DNA through a tiny protein pore (the nanopore), and ‘reading’ the DNA sequence base by base in real time, as the individual bases differentially affect the properties of the pore itself. Theoretically, the length of DNA string has no limit – as long as the DNA continues, the pore will keep reading.
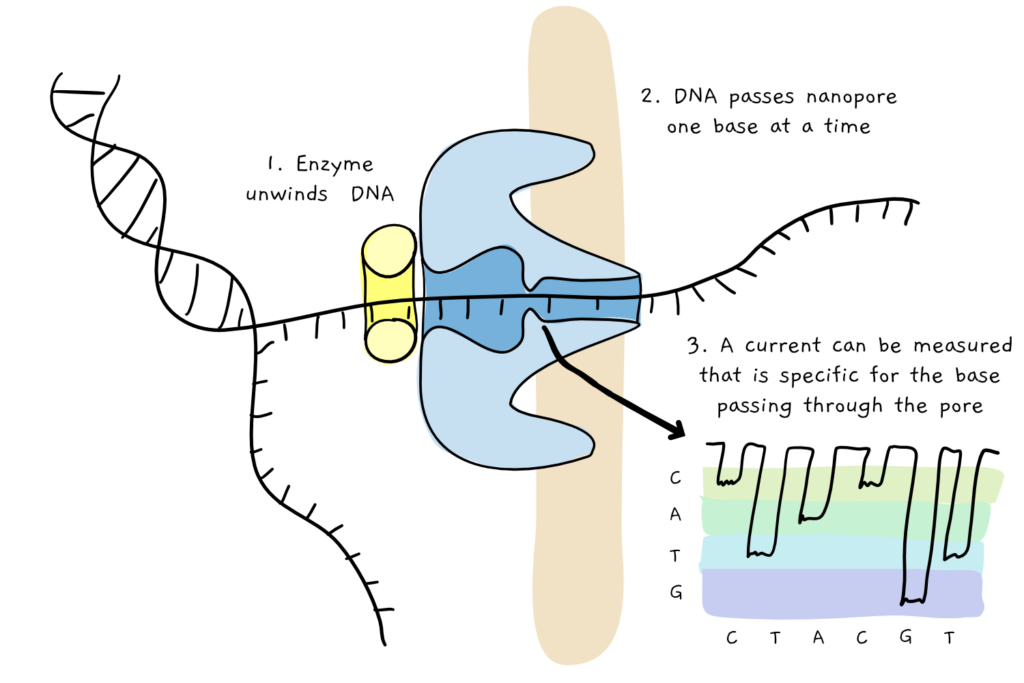
Queue a massive desire to find a way to extract looooooong DNA sequence. Apart from massively cutting down on the time it takes to compile (put together) all the short bits into one larger whole, having continuous long stretches of DNA can also help understand structural variations that could not be seen in shorter reads, including large deletions, inversions, and movement of genes around the genome. These, in turn, are particularly important in understanding how organisms evolve, as well as being involved in diseases such as cancer.
(As well as DNA, it can also read long stretches of RNA, which has massively helped us understand how organisms can take a single stretch of DNA, and use it to make a whole suite of different RNA molecules – like taking a box of lego pieces and building different types of structures).
Of course, we should note that nanopore sequencing is far from perfect: the major flaw is that the reads, while long, are pretty imprecise, with error rates (misreading or just missing a base) being upwards of 10%. These days, scientists tend to pair the longer less reliable nanopore sequencing with shorter but credible sequencing, to first build a (slightly shoddy) structure, and then put the correct sequences on top.
In the context of the extreme value of long reads and long read sequencing, and the amount of time and money that can be saved in data analysis, it’s not really surprising that scientists are more than happy to spend a couple of days and 50 USD or so on the DNA extraction iself. Zerpa-Catanho and colleagues’ method worked well on various plant species (although it was mostly tested on members of the pawpaw family, Caricaceae), produced large amounts of sequenceable DNA from only about 5 g of plant tissue, and resulted in DNA reads up to nearly 330,000 bases long.
The work included here shows us once more that scientific progress is not simply linked to an individual technology like nanopore sequencing, or the development of a single method. It might (not) shock you to hear that science is a team sport.
References:
Fun fact: long DNA fragments have been referred to as ‘whales’, with researchers promoting ‘whale spotting’ in their DNA data. Read more here:
Whales http://lab.loman.net/2017/03/09/ultrareads-for-nanopore/
A review of Nanopore sequencing functions; https://onlinelibrary.wiley.com/doi/full/10.1111/dgd.12608
Dessireé Zerpa-Catanho, Xiaodan Zhang, Jinjin Song, Alvaro G. Hernandez, Ray Ming, Ultra-long DNA molecule isolation from plant nuclei for ultra-long read genome sequencing, STAR Protocols, Volume 2, Issue 1, 2021, 100343,